
In a recent study published in Cell, Dr. Dinshaw Patel and Dr. Luciano Marraffini, along with their research team, unveiled a new mechanism by which bacteria use CRISPR systems to protect against viral invaders. The study, titled “The CRISPR-associated adenosine deaminase Cad1 converts ATP to ITP to provide antiviral immunity,” sheds light on a protein called CRISPR-associated adenosine deaminase 1 (Cad1). Patel is the Abby Rockefeller Mauze Chair of Experimental Therapeutics at Memorial Sloan Kettering Cancer Center, while Marraffini is the head of the Laboratory of Bacteriology at Rockefeller University.
The team’s findings provide a new understanding of how Type III CRISPR systems function, expanding the known range of immune strategies in bacteria beyond the usual nucleic acid degradation seen in other CRISPR defense mechanisms. Specifically, the study focuses on a protein domain within Cad1 called the CRISPR-associated Rossman fold (CARF), which activates antiviral processes through a cascade of cellular events triggered by binding molecules called cyclic oligoadenylates (cOAs). The Cad1 protein combines this CARF domain with an adenosine deaminase domain, which has the unique ability to transform ATP, the cell’s main energy molecule, into inosine triphosphate (ITP).
Upon recognizing a viral threat, a bacterial CRISPR system uses the Cas10 protein to create cOAs. This production of cOAs acts as an emergency signal to the CARF domain in Cad1, triggering it to bind these molecules and initiate ATP deamination. This deamination process, whereby Cad1 converts ATP to ITP, both halts bacterial cell growth and disrupts the life cycle of the infecting virus. The ability of Cad1 to shift cellular ATP to ITP probably represents a unique mode of antiviral defense that bacteria deploy to effectively combat viral infections.
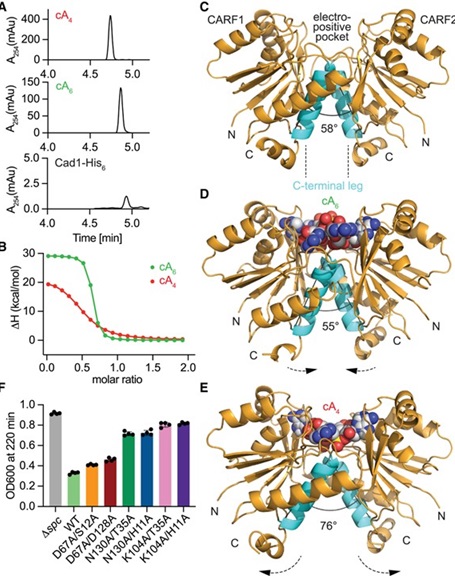
Using advanced cryoelectron microscopy (cryo-EM), the researchers unveiled the molecular architecture of Cad1. They discovered that the protein assembles into a six-unit structure, or hexamer, which positions the CARF and adenosine deaminase domains so as to maximize activity. The team’s structural studies revealed that, upon binding cOAs, Cad1 rearranges into a configuration that enhances its ability to convert ATP to ITP efficiently. This structure not only supports Cad1’s enzymatic function but also shows how ATP molecules bind within the protein’s inter-domain pockets, which is essential for triggering Cad1’s antiviral activity.
To study Cad1’s mechanism in action, the researchers introduced it into Staphylococcus aureus cells and observed how it responded to synthetic cOAs. They discovered that when activated, Cad1 rapidly converted ATP to ITP, causing a noticeable delay in bacterial growth without killing the cells. This reversible growth arrest, or “standby mode,” prevents the bacteria from becoming a viral replication factory, giving it a defense mechanism that stops viral spread within the bacterial population.
Further analysis demonstrated that Cad1’s transformation of ATP into ITP is directly linked to a specific cellular response to phage infection. The team explored Cad1’s interaction with different types of cOAs and found that it was primarily activated by cA4 and cA6, two specific cyclic molecules generated by the CRISPR system. Cad1’s ability to convert ATP to ITP, triggered by specific second messenger molecules, adds an additional layer of specificity to the CRISPR immune system. This discovery suggests that bacteria are equipped with a range of molecular tools to activate a precise and targeted response against phages.
In addition to providing an immediate growth arrest in the infected cells, Cad1’s unique method of ATP deamination to ITP may interfere with viral replication machinery, which requires ATP to function. By creating high levels of ITP, Cad1 likely disrupts viral propagation and curtails the infection’s spread within the host population.
Interestingly, this study revealed Cad1’s adaptability to different structural conformations, particularly the ability to recognize and bind either cA4 or cA6 ligands. This flexibility in ligand binding is significant because it suggests a modular adaptability of CRISPR systems in countering various viral infection strategies. The team’s experiments demonstrated that only the hexameric form of Cad1 is active in deaminating ATP, underscoring the structural necessity of this assembly for effective viral defense.
In testing Cad1’s effectiveness across various viral attacks, Patel and Marraffini’s team observed its immunity against a broad spectrum of phages that infect Staphylococcus bacteria. By pairing Cad1 with other CRISPR components, they found that the combination offered the highest level of immunity, even against viral attacks occurring later in the infection cycle.
This discovery has important implications for understanding the evolutionary pressures shaping bacterial immune systems. These findings reveal how CRISPR systems continue to evolve new strategies, like Cad1’s ATP deamination, to respond to the diverse threats posed by phages. The research hints that the evolutionary arms race between bacteria and their viral attackers drives the development of complex CRISPR immune responses, helping bacteria survive in increasingly hostile environments.
Despite its findings, the study does acknowledge limitations. For example, the team expressed caution in generalizing the exact behavior of Cad1 in all bacterial systems, as their experiments utilized a specific Staphylococcus strain. Further studies may explore the nuances of Cad1 in different bacterial hosts, as well as the exact molecular pathway by which Cad1 shifts into a catalytically active state upon cOA binding.


